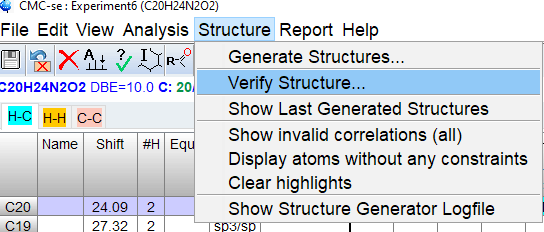
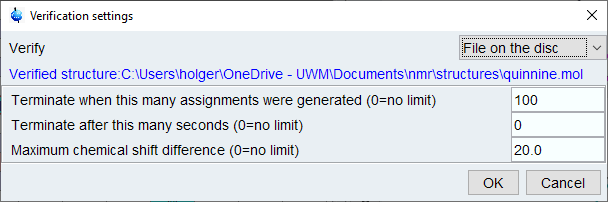
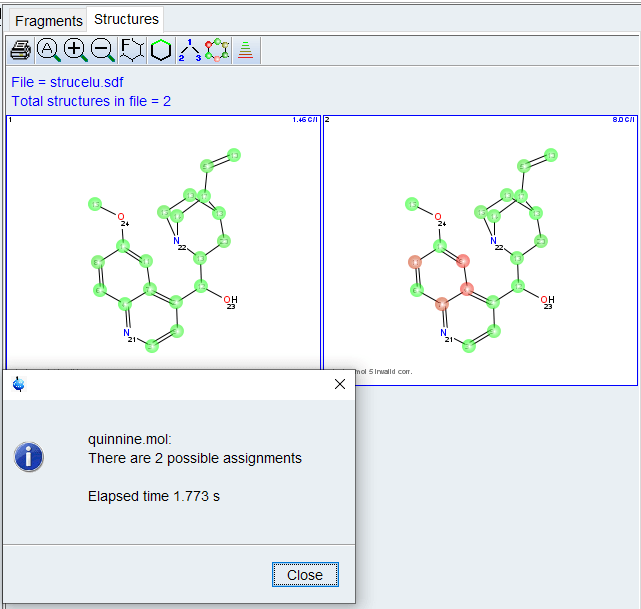
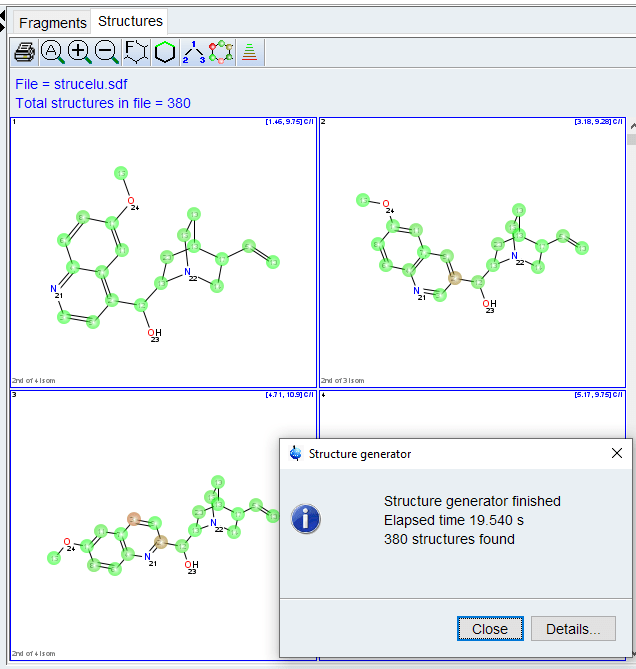
General
Remote operation of the spectrometer gives you the same control you have when physically sitting at the spectrometer computer. There is no need to be logged in and have ICONNMR running prior to remote access as required for the ICONNMR based web server. Only your samples need to be placed in the sample changer or inserted in the magnet ahead in time. So you can prepare samples during the day, leave them in the sample changer and run them later from your office or from home, or you can check on an experiment started earlier.
Off Campus Users
Access from outside the UWM campus requires the user to be connected to the UWM VPN (Virtual Private Network). Please follow the links below how to install and run the Global Protect VPN software.
Installing GlobalProtect:
https://kb.uwm.edu/83286 (Windows) https://kb.uwm.edu/83289 (Mac)
Connecting to VPN:
https://kb.uwm.edu/89581(Windows) https://kb.uwm.edu/89582
Running Windows Remote Desktop Client
The remote desktop client is part of the standard Windows 10™ installation. It can be accessed from the Windows Accessories in the Start menu.

You can also find it be typing “Remote” into the search bar.

Connecting With Remote Desktop Client
In the main Remote Desktop window, enter the full host name of the spectrometer. For the Avance 500 instrument, that is nmr500.chem.uwm.edu, for the 400MHz spectrometer it is nmr400.chem.uwm.edu. Then click the [Connect] button.
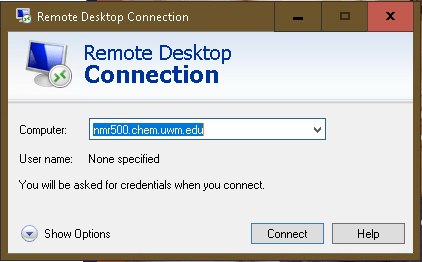
Expanding on “Show Options” allows to save your username in a profile and pre-define window sizes. Full Screen is recommended.


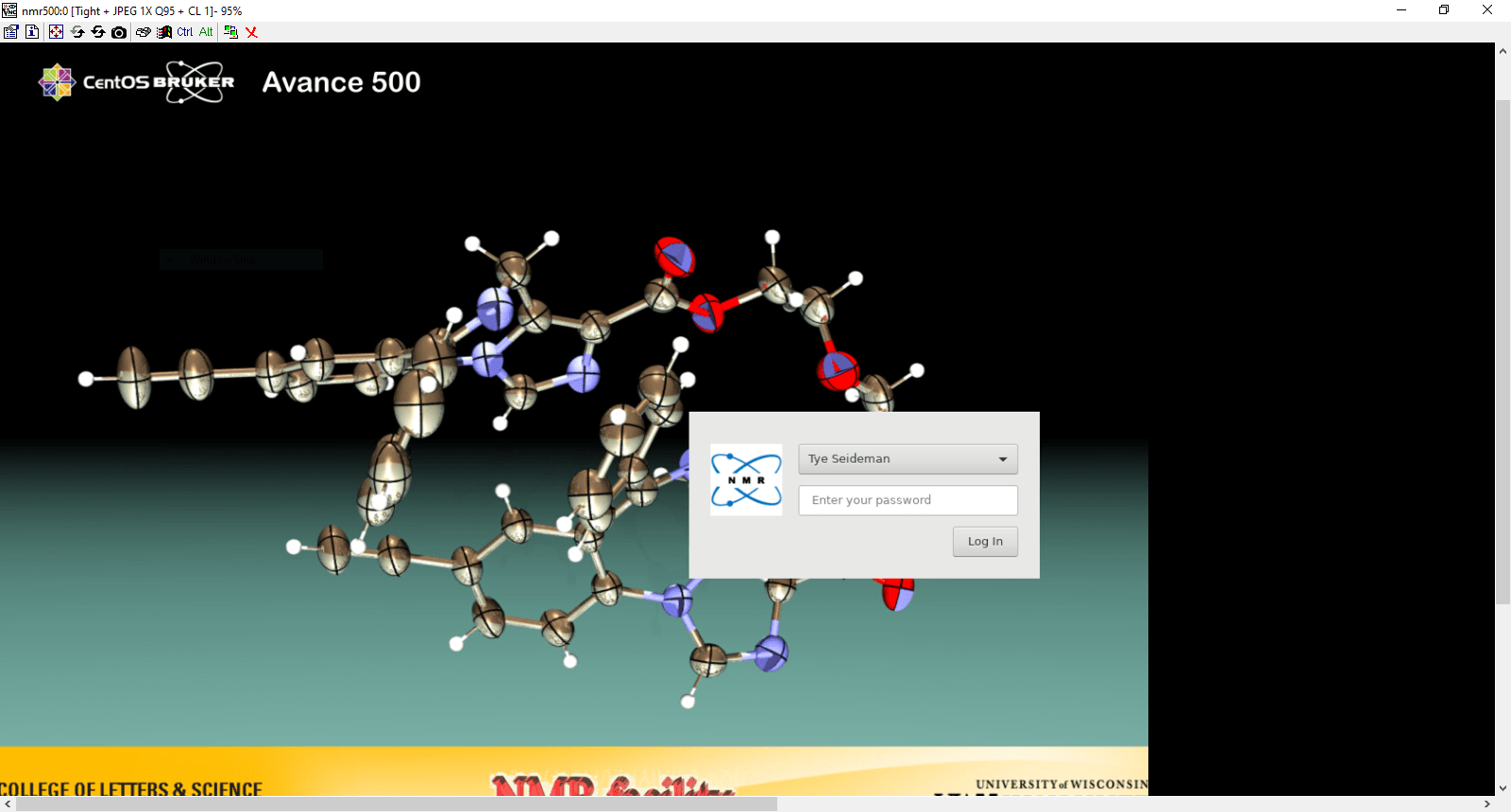
In the server login window, keep the default session “Avance 500 Main Desktop” and enter your username and password (Username may already be filled in if entered in the previous window.
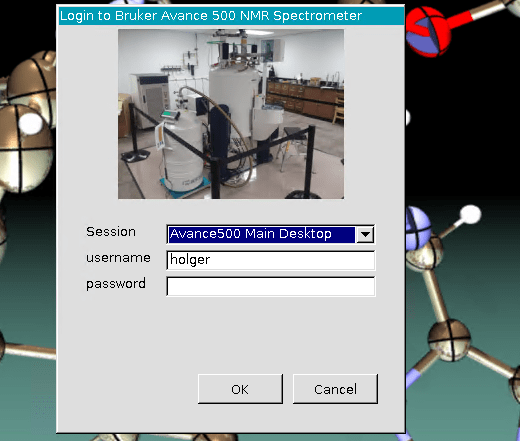
This will log you into the desktop currently running on the spectrometer. If nobody is logged in, that will give you the main login screen as you would see when in front of the computer directly. This behavior is different from connecting to a Windows computer, where you automatically lg into a new session.
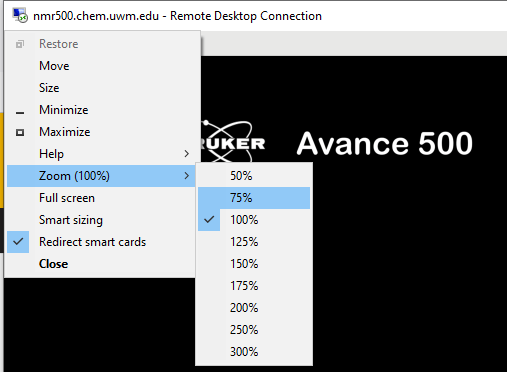
If the remote screen is larger than yours, you may need to scroll to find the relevant part of your screen. Unfortunately, as opposed to the Turbo VNC viewer, zooming of the display is not possible in full screen mode.

Zooming is possible in window mode. Exit full screen mode by clicking on the restore windows icon on the tab on top of the window.
![]()
Then, click on the icon in the top left corner to access display options. Note that on small monitors zooming out may give you the full screen, but at the cost of readability. Returning to Full screen is only possible after setting zoom back to 100%.