The procedure for running two dimensional spectra follows in general the procedure for one dimensional experiments with some modifications.
Preparatory experiments
It is recommended to always take a proton spectrum to verify sample purity, assess concentration and later use to optimize spectral window.
A one dimensional carbon spectrum can also be useful, but not essential and may not always be feasible.
It is easiest if the 1D experiments are stored under the same name as the 2D experiments.
Select experiment
During creation of dataset, select the 2D experiment of choice. Predefined parameter files exist for most common two dimensional experiments:
- 1H/1H experiments:cosy, dqfcosy, tocsy, noesy, roesy
- 1H/13C experiments:c13hsqc, c13hsqced, c13hmbc, c13adequate, c13hsqctocsy
- other nuclei: n15hsqc, n15hmbc,si29hmbc
Setting up an experiment usually involves defining 1D proton and heteronuclear experiment (if available) for projection, optimizing the specral width in both dimensions, and adjusting the duration of the experiment for sign al to noise and sensitivity.
Tuning/Matching
Proper tuning/matching of the probe is usually required. It needs to be performed only once for each sample, but for all nuclei used in the experiment (1H and C13 for HSQC.
Turn rotation OFF
Spinning a sample can improve the line width, but also introduces diffusion and instability into the sample. 2 D experiments are typically run without sample rotation:
Type ro off on the command line or select from [Spin] menu:

Interactive optimization of spectral width
This procedure uses existing 1D proton (and carbon if applicable) spectra to interactively set spectral windows. The spectra will also be added as 1D projection for plotting.
- Select [Set Limits] from the Acquire flow bar. A text box with step by step instructions will appear
![]()

- Call up the 1D proton spectrum. If you stored it with the same dataset name, just read the respective experiment number using the command line, i.e. re 1
- You can also use the browser or the data history

- Zoom the region you want to acquire in the 2D spectrum, then click [Ok] in the instructions dialog. The software will automatically select the proper dimension.


- Repeat the process for carbon (or other hetero nucleus) if applicable.
Manually setting spectral width
If no 1D spectrum is available, you can manually set spectral window parameters by typing o1p (offset in ppm) and sw (spectral width) at the command line. The parameter for the acquisition dimension (horizontal, typically 1H) is on the left, the indirect dimension (vertical, can be 1H, carbon or other X nucleus) is on the right.
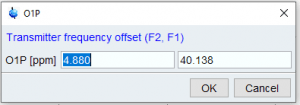

- Setting o1p in the indirect dimension for a HSQC will also set the dependent parameter o2p
- Toe achieve a window leftppm to rightppm you need to set you need to set sw = leftppm – rightppm and o1p = rightppm + sw/2
Setting experiment time
Default parameters for number of scans (ns) and number of individual spectra (1td) work for most samples. If your sample is dilute or you need extra resolution in the indirect dimension, you may need to extend the experiment time.
- ns increases the number of scans per spectrum. That will increase signal to noise, but not change the resolution in the indirect dimension
- 1 td will increase the total number of experiments done for the 2D. This will typically improve both resolution and signal to noise
- When typing td, only change number of points in the indirect dimenstion (f1, right value)
