Phasing Methods for the Structure Completion Problem
Proteins form the basic building blocks of all components of living organisms, from their muscles to the enzymes that catalyse vital chemical reactions. Proteins are created from instructions coded in DNA. Since the recent completion of the human genome project, essentially the entire genetic map of humans is known, and hence via the genetic code, the amino acid sequence of all human proteins.
The function of a protein is largely a consequence of its three-dimensional shape, which is determined from its peptide sequence by the still unknown mechanisms of protein folding. In practice, the 3D structures of proteins are still largely discovered by the technique of x-ray crystallography. In this technique proteins are coaxed into ordered arrays, or crystals, and the structure of each repeat unit (or unit cell) is determined by an analysis of the intensities of the Brag relections of incident x-rays.
The major obstacle to solving the structure of a protein from such diffraction data is the phase problem. In protein crystallography the process of solving this problem usually begins with the estimation of approximate phases by one of the following three techniques: (a) the isomorphous replacement method, which requires the preparation of related structures with heavy atoms attached to the original protein, (b) the multiple-wavelength anomalous-dispersion (MAD) method, which depends on the presence of sufficiently strong anomalously scattering atoms within the protein, and (c) the molecular replacement method, which requires the identification of a known structure similar to the one whose structure is sought. Initial phases obtained by any of these methods must be refined for a successful structure determination. After an interpretable electron density map is obtained, an initial atomic model is built and subsequently refined against the experimental data.
When a partial model of a protein has initially been determined by any of these methods, it is often possible to determine the rest of the structure by the process of structure completion. The most basic of these methods are the class known as difference Fourier methods (W. Cochran, Acta Cryst. 4, 408 (1951))
When the known part of the structure is not a major part of the molecule, the difference Fourier method may become unreliable. The methods we have developed are much more accurate. This is demonstrated by the example below from our paper, D. K. Saldin, V. L. Shneerson, and D. L. Wild, J. Imaging Sci. Technol., 41, 482-487 (1997), on the protein bovine pancreatic trypsin inhibitor (BPTI). A “known” molecular fragment was created by deleting from the full structure amino acid residues 1-28 (approx. 50% of the molecule). We then attempted to recover the deleted residues from just the Bragg intensities from the full structure and knowledge of the “known” fragment.
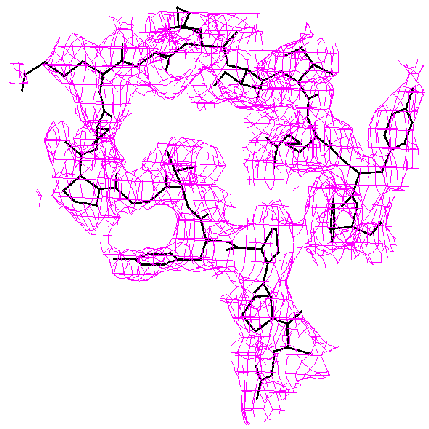 |
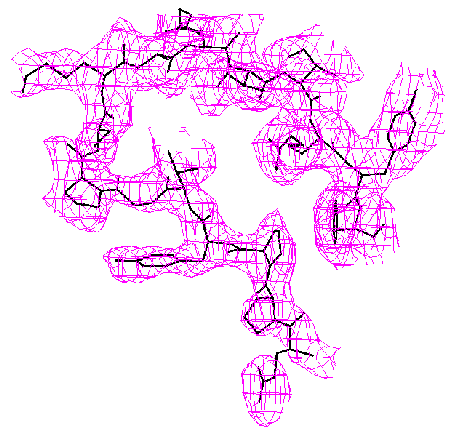 |
| Difference Fourier Reconstruction Reconstruction by our Iterative Phasing Algorithm The figure above shows an isosurface of the missing electron density of residues 7-22 of BPTI as recovered by the difference Fourier method. The true structure is indicated by the stick figure. Some false connectivity and other incorrect electron density reconstruction is apparent. |